Have you kept your ear to the ground? Felt something on the breeze? Getting a "gut feeling"?
The most recent edition of Chemistry Bumper Cars - Faculty Moves, for the uninitiated - leans towards bigger deals and dramatic poaches as the Fall term looms over the Summer horizon. Here's the latest I've heard about, with my own opinion about whether the rumor holds water.
Dave MacMillan to leave Princeton, for...?
Odds: Low
I hear what you're saying: MacMillan has already moved twice (Berkeley -> Caltech -> Princeton), and we're talking about a researcher who averages an award every year and a new named professorship every four. However, he's fairly well settled into a tight relationship with Merck, who are local to NJ. He's also helped propel Princeton back up in the rankings over the past decade. I can think of only one university that sounds like any kind of a step up, and they have plenty of organic power at the moment.
Dirk Trauner to NYU
Odds: Medium
Though I've heard this more than once, I'm scratching my head about how it makes sense for Trauner. Part of his motivation in returning to LMU was to continue the Mulzer mystique: the powerhouse European natural products group that makes densely-functionalized products appear as if by magic. Then again, NYU seems to be aggressively searching for a certain kind of chemist; maybe Dirk is slated to be the new Phil Baran of the East Coast?*
Update: As seen in the comments, Dirk himself confirms. My gracious thanks to the Professor.
Tom Rovis to Columbia
Odds: Certain
Signed, sealed, and delivered to Columbia back in the Spring.
Dave Liu to Broad from Harvard
Odds: Low
First he was an undergraduate wunderkind with Corey, now one of the youngest Full Professors and an HHMI scholar, all before age 40. He's already a core faculty member with Broad while managing his Harvard group, and I see no reason for Harvard (or for Liu) to wish to terminate his current position. This may sound like wild speculation or stargazing, but I fully suspect Liu's name goes on a nomination for a Big Prize within ~5 yrs, and I think Harvard would do everything they could to keep him in the fold for that day.
Update: As noted in the comments, does appear Liu will have to be physically present on the Broad's campus.
Karen Goldberg to leave U. Washington
Odds: Low
I very much want to believe, especially since UW lost Jim Mayer a few years back, that they can retain Goldberg, a C-H activation and general OM superstar. She boasts a local Center and a named professorship, as well as a Department with plenty of talented young blood: Boydston, Bush, Cossairt, Fu, Lalic, Schlenker, Theberge, Zalatan, all hired in just the last 6 years, doubtless some drawn there through her influence. I'm sure she'd succeed at a Caltech or an MIT, but I really don't know enough about her motivations to say any more conclusively.
Greg Verdine leaves Harvard to run companies full-time
Odds: High
It's said you can throw a rock in Cambridge these days and hit a VC. Seeing how much apparent fun and success Verdine has had with his previous ventures into the private sector, I'm betting he continues this line full-time and slowly winds down managing theses and group meetings.
--
*Today's ridiculous statistic: In the past 20 years, Baran and Trauner have authored a combined 372 research papers. That's 2-3 entire careers, and these are guys with 20+ years ahead of them. Damn.
Showing posts with label catalysis. Show all posts
Showing posts with label catalysis. Show all posts
Saturday, July 9, 2016
Monday, June 29, 2015
(Please) Make More Molecules using Light!
Update (6/29) - Commenters chime in with some notables I'd mistakenly left off the list. I'll append their molecules to the end of this post (vide infra)...
I'm officially calling it: Photoredox catalysis = the new "it reaction" for organic chemistry.
Like many before it - iron catalysis, the gold rush, anything palladium, organocatalysis - photoredox catalysis is now appearing in my RSS feed on a near-hourly basis. We're in the early days of an exciting field; I've noticed more methodology papers and mechanistic studies lately. Assuming you start with a suitable aryl halide, diazonium, or carboxylic acid, the synthetic toolkit of single-electron catalysis seems virtually limitless.
Which begs the question . . .where are all the photoredox total syntheses?
SciFinder: "photoredox total synthesis?"
7 hits.
How about "light-mediated total synthesis?"
7 more.
One more try: "photoredox natural products?"
7 hits.
Most of these hits actually lead to conference abstracts, not individual manuscripts. Props to the Stephenson group (Michigan), who leads the charge with syntheses of aspidosperma alkaloids and gliocladin C. Based on SciFinder results, I'd include the MacMillan synthesis of Lyrica, and Lei's isoquinoline syntheses from JOC.
Readers, what am I missing? A pivotal review or book? A group whose research is at the forefront of solar-powered natural product production? Perhaps a major non-English journal article? Any examples where a venerable old lion of total synthesis utilized a photoredox reaction alongside their Diels-Alders and aldol reactions?
For such a large-upside field, it sure seems quiet out there.
--
Update: Molecules made with photoredox catalysis, as suggested by my beloved commenters:
Overman, (-)-aplyviolene, Ru(bpy)3
MacMillan, fenofibrate (OK, not a np), Ni(II), Ir(III)
MacMillan, (-)-burshernin, Ru(bpy)3
Nicewicz, methylenolactocin and Protolichesterinic Acid, acridinium
Nicewicz, magnosalin + pellucidin A
Yoon, heitziamide A, Ru(bpz)3
Yoon, epiraikovenal, Ir(III)
Chen + Baran, sceptrin, Ir(ppz)3
Chen, nakamuric acid, Ir(ppy)3
Lawrence / Sherburn, endiandric acid A, kingianins A,D,F, kingianic acid E, Ru(bpy)3
Carreira, (+)-Daphmanidin E, Co-diimine (method here)
More?
I'm officially calling it: Photoredox catalysis = the new "it reaction" for organic chemistry.
Like many before it - iron catalysis, the gold rush, anything palladium, organocatalysis - photoredox catalysis is now appearing in my RSS feed on a near-hourly basis. We're in the early days of an exciting field; I've noticed more methodology papers and mechanistic studies lately. Assuming you start with a suitable aryl halide, diazonium, or carboxylic acid, the synthetic toolkit of single-electron catalysis seems virtually limitless.
Which begs the question . . .where are all the photoredox total syntheses?
 |
| Three examples of recent total syntheses that capitalize on photoredox catalysis (bonds in red) |
7 hits.
How about "light-mediated total synthesis?"
7 more.
One more try: "photoredox natural products?"
7 hits.
Most of these hits actually lead to conference abstracts, not individual manuscripts. Props to the Stephenson group (Michigan), who leads the charge with syntheses of aspidosperma alkaloids and gliocladin C. Based on SciFinder results, I'd include the MacMillan synthesis of Lyrica, and Lei's isoquinoline syntheses from JOC.
Readers, what am I missing? A pivotal review or book? A group whose research is at the forefront of solar-powered natural product production? Perhaps a major non-English journal article? Any examples where a venerable old lion of total synthesis utilized a photoredox reaction alongside their Diels-Alders and aldol reactions?
For such a large-upside field, it sure seems quiet out there.
--
Update: Molecules made with photoredox catalysis, as suggested by my beloved commenters:
Overman, (-)-aplyviolene, Ru(bpy)3
MacMillan, fenofibrate (OK, not a np), Ni(II), Ir(III)
MacMillan, (-)-burshernin, Ru(bpy)3
Nicewicz, methylenolactocin and Protolichesterinic Acid, acridinium
Nicewicz, magnosalin + pellucidin A
Yoon, heitziamide A, Ru(bpz)3
Yoon, epiraikovenal, Ir(III)
Chen + Baran, sceptrin, Ir(ppz)3
Chen, nakamuric acid, Ir(ppy)3
Lawrence / Sherburn, endiandric acid A, kingianins A,D,F, kingianic acid E, Ru(bpy)3
Carreira, (+)-Daphmanidin E, Co-diimine (method here)
More?
Wednesday, June 17, 2015
Lord of the (Small) Rings
Quick: What small, odd-looking thing carries metal through harsh trials?
Here's hoping you answered the Doyle group's new ligand: "Fro-DO."
Unlike common sigma donors - NHCs, amines, phosphorous ligands - EDO (electron-deficient olefin) ligands function as pi-acceptors. Instead of dumping electron density into oxidative addition (adding an R-X bond across a metal atom) EDOs speed up* the other side of catalysis, namely reductive elimination (joining the organic fragments and restoring the metal's electrons). According to the authors, acceleration of reductive elimination helps to decrease the amount of substrate decomposition due to beta-hydride elimination.

Doyle and coworker Dennis Huang report selective ring-opening of aziridines - no mean feat in itself - and generate a quaternary center in the process, in 31-86% yields. Using a modified camphor-like sultam for their EDO, the group observes 27% ee, sure to be the focus of its own publication in the near future. Curious about related efforts in other groups? I recommend this Jamison mini-review.
Now, back to the name: "Fro-DO" carries the torch for a small-but-growing literature subculture. Chalk up another example to what The Atlantic recently called "Science's Love Affair with LOTR." I spent a few moments with SciFinder, trying to dig up some more chemistry-themed examples; the Atlantic points out many from geology and astronomy, and precious few from our molecular audience. Without further ado:
Here's hoping you answered the Doyle group's new ligand: "Fro-DO."
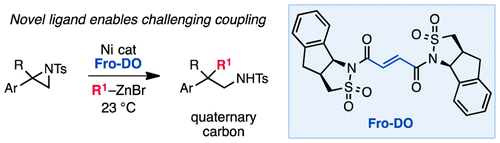 |
| I see what you did there. Credit: Doyle group, JACS 2015 |
Unlike common sigma donors - NHCs, amines, phosphorous ligands - EDO (electron-deficient olefin) ligands function as pi-acceptors. Instead of dumping electron density into oxidative addition (adding an R-X bond across a metal atom) EDOs speed up* the other side of catalysis, namely reductive elimination (joining the organic fragments and restoring the metal's electrons). According to the authors, acceleration of reductive elimination helps to decrease the amount of substrate decomposition due to beta-hydride elimination.

Doyle and coworker Dennis Huang report selective ring-opening of aziridines - no mean feat in itself - and generate a quaternary center in the process, in 31-86% yields. Using a modified camphor-like sultam for their EDO, the group observes 27% ee, sure to be the focus of its own publication in the near future. Curious about related efforts in other groups? I recommend this Jamison mini-review.
Now, back to the name: "Fro-DO" carries the torch for a small-but-growing literature subculture. Chalk up another example to what The Atlantic recently called "Science's Love Affair with LOTR." I spent a few moments with SciFinder, trying to dig up some more chemistry-themed examples; the Atlantic points out many from geology and astronomy, and precious few from our molecular audience. Without further ado:
- Superconducting magnets used in fusion research, controlled by codes nicknamed SARUMAN and GANDALF
- Retrotransposons in a .small flowering plant, dubbed "Gimli," "Gloin," and "Legolas"
- A breast cancer gene marker, called "Frodo"
- MRI pulse sequences, used to eliminate artifacts, also dubbed "FRODO"
- Finally, a docking program for small molecules and RNA, with an apt name: "MORDOR"
Readers, any more chemistry-themed LOTR callbacks? Send 'em along!
--
*...or maybe not. An observant commenter on Reddit noticed that Doyle and colleagues see no correlation between 13C shift of the olefinic EDO carbons and reaction rate. They posit, instead, substantial steric congestion around the metal surface as responsible for the rate enhancement.
--
*...or maybe not. An observant commenter on Reddit noticed that Doyle and colleagues see no correlation between 13C shift of the olefinic EDO carbons and reaction rate. They posit, instead, substantial steric congestion around the metal surface as responsible for the rate enhancement.
Thursday, April 3, 2014
Gold Steps Up
Update, 4/5/14: On Twitter, Yunus chimes in to recommend this Hashmi paper, showing a mononuclear Au(I) catalyst - with crazy adamantyl appendages - that gets down to 0.0001% loading!
--
Catalyst: A substance that increases the rate of a reaction without modifying the overall standard Gibbs energy change in the reaction (IUPAC Gold Book).
Though most chemists agree on the above definition, there have always been two camps: "Academic" catalysis (1-20 mol%, 5-100 turnovers), and "Industrial" Catalysis (<0.01 mol%, >10,000 turnovers). As more researchers seek to translate early discoveries into efficient, "green" processes, the desire for robust organometallic species has skyrocketed.
 Right now, a few such homogeneous catalysts have made the jump: the Grela Ru metathesis catalysts (0.001%), copper catalysts for boron addition (0.005%), and some iridium and palladium species.
Right now, a few such homogeneous catalysts have made the jump: the Grela Ru metathesis catalysts (0.001%), copper catalysts for boron addition (0.005%), and some iridium and palladium species.
Gerald Hammond and Bo Xu (Louisville) want to add one more to the list: a highly-stable gold catalyst. Their new catalyst, dubbed "BisPhePhosXD-AuCl" (Phew!), takes development lessons from Buchwald (electron-rich, C-to-metal bond) and Widenhoefer (steric bulk to discourage off-cycle species).
Now here's the fun part: the authors run several typical gold-catalyzed reactions side-by-side with their improved catalyst. In most cases, they're able to shave off 99.9% of the catalyst loading, with comparable yields. Granted, certain reactions take more time with this approach, but a few finish ahead of their literature counterparts. Wow.
Aldrich sells the precatalyst, if you're itching to try it yourself. I'll be very interested to see the next generation of bulked-up ligands, and whether this reactivity transfers over to other precious metals, namely platinum, iridium, or rhodium.
--
Catalyst: A substance that increases the rate of a reaction without modifying the overall standard Gibbs energy change in the reaction (IUPAC Gold Book).
Though most chemists agree on the above definition, there have always been two camps: "Academic" catalysis (1-20 mol%, 5-100 turnovers), and "Industrial" Catalysis (<0.01 mol%, >10,000 turnovers). As more researchers seek to translate early discoveries into efficient, "green" processes, the desire for robust organometallic species has skyrocketed.
Right now, a few such homogeneous catalysts have made the jump: the Grela Ru metathesis catalysts (0.001%), copper catalysts for boron addition (0.005%), and some iridium and palladium species.Gerald Hammond and Bo Xu (Louisville) want to add one more to the list: a highly-stable gold catalyst. Their new catalyst, dubbed "BisPhePhosXD-AuCl" (Phew!), takes development lessons from Buchwald (electron-rich, C-to-metal bond) and Widenhoefer (steric bulk to discourage off-cycle species).
Now here's the fun part: the authors run several typical gold-catalyzed reactions side-by-side with their improved catalyst. In most cases, they're able to shave off 99.9% of the catalyst loading, with comparable yields. Granted, certain reactions take more time with this approach, but a few finish ahead of their literature counterparts. Wow.
Aldrich sells the precatalyst, if you're itching to try it yourself. I'll be very interested to see the next generation of bulked-up ligands, and whether this reactivity transfers over to other precious metals, namely platinum, iridium, or rhodium.
Tuesday, April 1, 2014
"Everything is Catalytic," Scientists Claim
For Immediate Release
4/1/14
Grand Rapids, MI: Troublesome chemical reactions? Try adding a pinch of...anything.
Reporting today in the journal ACS Catalysis, researchers have discovered that every chemical element or molecular mixture catalyzes reactions when present in trace quantities. "As I've told all my students, catalytic inspiration + 10 equivalents perspiration produces beautiful molecules," remarked Scripps Professor Phil Baran. "I just never mentioned that I used drops of actual sweat!"
"Brilliant!" remarked Stuart Cantrill, Chief Editor of Nature Chemistry. "Chemists were always running reactions in beer and coffee, mostly to show off. The trick now will be discovering which obscure thing goes into what reaction."
"Indeed," remarked Chemistry World's Neil Withers.

As shown by the graphic abstract (above), scientists at the forefront of catalytic research often try just about anything they can get their hands on. "I wouldn't have believed it, myself, but the data convinced me," commented celebrated catalysis scion John Hartwig. "Our lab has already added ppm quantities of dryer lint, nose hairs, and soy sauce to asymmetric Ir allylations, with fantastic yields and high ee."
N.B. - Calls to Dow and DuPont were not returned by press time
Note to the humorless: This is fake. Happy April Fools' Day. Please don't sue me.
4/1/14
Grand Rapids, MI: Troublesome chemical reactions? Try adding a pinch of...anything.
Reporting today in the journal ACS Catalysis, researchers have discovered that every chemical element or molecular mixture catalyzes reactions when present in trace quantities. "As I've told all my students, catalytic inspiration + 10 equivalents perspiration produces beautiful molecules," remarked Scripps Professor Phil Baran. "I just never mentioned that I used drops of actual sweat!"
"Brilliant!" remarked Stuart Cantrill, Chief Editor of Nature Chemistry. "Chemists were always running reactions in beer and coffee, mostly to show off. The trick now will be discovering which obscure thing goes into what reaction."
"Indeed," remarked Chemistry World's Neil Withers.
As shown by the graphic abstract (above), scientists at the forefront of catalytic research often try just about anything they can get their hands on. "I wouldn't have believed it, myself, but the data convinced me," commented celebrated catalysis scion John Hartwig. "Our lab has already added ppm quantities of dryer lint, nose hairs, and soy sauce to asymmetric Ir allylations, with fantastic yields and high ee."
N.B. - Calls to Dow and DuPont were not returned by press time
Note to the humorless: This is fake. Happy April Fools' Day. Please don't sue me.
Sunday, March 16, 2014
'March Madness' - Cobalt Catalysis?
Catalysis fads come in waves. This shouldn't surprise - when one group finds almost-too-good-to-be-true reactivity, everyone jumps on to ensure rapid publication and novelty for that dusty grant submission.
For precious metals, King Palladium still reigns supreme, though some serious recent coups came courtesy of gold, iron, rhodium, and iridium. But a new challenger now looms on the horizon:
cheap, plentiful COBALT.

Within about a week of one another, the Dong, Chirik, and Yoshikai groups have disclosed some really neat-o transformations that run on the Co(I)-Co(III)* redox engine. Dong's group discovered a new diene hydroacylation. Chirik's installed B(pin) onto heteroaromatics with low loading and no solvent. Yoshikai offers a mini-career-retrospective on his group's efforts to develop several (new to me!) Co reactions, among them hydroarylation, zinc insertion, and acylation.
So, why the sudden upswing in cobalt? For one, relatively new ligand architectures (pincers, NHCs) have allowed access to stable architectures earlier chemists could only dream of. Also, the concept of "reductive" coupling, still in its infancy compared to the oxidation behavior most folks affiliate with top-row metals, has lured younger groups fighting to carve out a career niche.
Let's see how swollen the cobalt catalysis field becomes in the next five years. The next "gold rush?"
Only time will tell.
*As @Organometallica (rightly) points out, these reactions may yet be more complex - say, for instance, a fast Co(I)-Co(II) to Co(II)-Co(III). This could have mechanistic ramifications later down the road (radicals, anyone?), or be responsible for various off-cycle activity / resting states.
For precious metals, King Palladium still reigns supreme, though some serious recent coups came courtesy of gold, iron, rhodium, and iridium. But a new challenger now looms on the horizon:
cheap, plentiful COBALT.
Within about a week of one another, the Dong, Chirik, and Yoshikai groups have disclosed some really neat-o transformations that run on the Co(I)-Co(III)* redox engine. Dong's group discovered a new diene hydroacylation. Chirik's installed B(pin) onto heteroaromatics with low loading and no solvent. Yoshikai offers a mini-career-retrospective on his group's efforts to develop several (new to me!) Co reactions, among them hydroarylation, zinc insertion, and acylation.
So, why the sudden upswing in cobalt? For one, relatively new ligand architectures (pincers, NHCs) have allowed access to stable architectures earlier chemists could only dream of. Also, the concept of "reductive" coupling, still in its infancy compared to the oxidation behavior most folks affiliate with top-row metals, has lured younger groups fighting to carve out a career niche.
Let's see how swollen the cobalt catalysis field becomes in the next five years. The next "gold rush?"
Only time will tell.
*As @Organometallica (rightly) points out, these reactions may yet be more complex - say, for instance, a fast Co(I)-Co(II) to Co(II)-Co(III). This could have mechanistic ramifications later down the road (radicals, anyone?), or be responsible for various off-cycle activity / resting states.
Friday, February 21, 2014
Rhodium Gets It Done
 |
| Interesting, informative intermediates from Rh(I) silylation |
The authors quickly point out that this silylation runs at "low" temps (80 deg C), uses fairly cheap commercially-available reagents, and occurs with almost reversed selectivity to the corresponding direct borylation conditions. But my favorite part comes from a deep dive into the Supporting Information. Far from the discussion of academic over-publishing we've had for the past few days, Hartwig and Cheng really sculpt a great paper here: Stability studies. Reactivity differences (Si vs. B). Cross-couplings. Preliminary mechanistic details.
As always, there's tons more to do. Getting out of the glovebox with a more stable Rh precursor, or translating the reactivity to an earlier metal (a tall order!). Deeper mechanistic studies would certainly show the way. Even more tantalizing? Using single-enantiomer versions of the bulky ligands to incorporate some chiral-at-silicon synthons. I can't wait to see the rest of this story.
Thursday, February 13, 2014
Cyclobutanone Déjà vu
Didn't I just see that reaction? [rubs eyes]
From the laboratory of Prof. Nicolai Cramer (EPF Lausanne) comes some really neat examples of cyclobutanone C-C activation. Just mix with ~2 mol% of cationic Rh(I), and presto! Out comes a [3.1.2] bicyclo product reminiscent of several neuroactive natural products.
Oftentimes, a catalytic reaction will go gangbusters, but will stubbornly refuse all attempts at asymmetric induction. Apparently not so here - Cramer reports the asymmetric version in ACIEE about two weeks after the initial Organometallics report!
Honestly, I might never have noticed, except the TOC graphics for both are nearly identical: a comic pair of orange scissors "snipping" apart the cyclobutanone ring.
So, readers, what's your take? Can't wait to see this reaction applied to a challenging target? Or, a strange choice of synthetic "least publishable units" (LPUs)?
From the laboratory of Prof. Nicolai Cramer (EPF Lausanne) comes some really neat examples of cyclobutanone C-C activation. Just mix with ~2 mol% of cationic Rh(I), and presto! Out comes a [3.1.2] bicyclo product reminiscent of several neuroactive natural products.
Oftentimes, a catalytic reaction will go gangbusters, but will stubbornly refuse all attempts at asymmetric induction. Apparently not so here - Cramer reports the asymmetric version in ACIEE about two weeks after the initial Organometallics report!
 |
| Angew. Chem. Int. Ed. 2014, ASAP |
 |
| Organometallics 2014, ASAP |
So, readers, what's your take? Can't wait to see this reaction applied to a challenging target? Or, a strange choice of synthetic "least publishable units" (LPUs)?
Thursday, August 29, 2013
Bruceollines: Short and Sweet
You can't put your finger on it, but sometimes you just feel compelled to read a paper. This one, from Org. Lett. ASAP, scratched all the usual itches: protecting-group free total synthesis (check), traditional medicine (check), tropical diseases (check), and cool off-the-shelf reagents (check).
I must admit, Gordon Gribble's name at the top caught my attention, but his co-authors hail from the University of the West Indies, a school I've always been curious about.*
The goal? Fast, selective production of bruceollines, medicinal compounds isolated from the roots of a Chinese shrub. The authors initially try to assemble the common indole (the 6-5 aromatic ring) core of the bruceolline family using Fischer conditions, but the starting materials have other ideas and form non-productive intermediates. Starting over, palladium catalysis proceeds smoothly, then relatively gentle oxidation (DDQ) produces the fully oxidized bruceolline E (see picture).

To access the final compound, Gribble & Co. must reduce just one of E's two ketones. The authors attempt borane reductions, but the "usual suspects" (CBS, Alpine) fail miserably. Optimization with (+)-DIP-Cl produces the final bruceolline J in high yield and ee. To make the unnatural enantiomer, the authors turn to a personal favorite: Baker's yeast, more commonly found in breads than labs. After 14 days in a warm, sweet slurry, the wee beasties return ent-bruceolline J in 98% ee.
The synthesis, only 4 short steps, should open the door to develop new antimalarial compounds.
*First, the Hawaii paradox - recruiting high-end, serious grad students to work 4-6 years in a tropical paradise. How does that work? And how do shipping delays from the mainland impact project selection? I can imagine that protecting group-free, relatively robust chemistry would have to be the norm, to survive storms, delays, humidity, etc.
I must admit, Gordon Gribble's name at the top caught my attention, but his co-authors hail from the University of the West Indies, a school I've always been curious about.*
The goal? Fast, selective production of bruceollines, medicinal compounds isolated from the roots of a Chinese shrub. The authors initially try to assemble the common indole (the 6-5 aromatic ring) core of the bruceolline family using Fischer conditions, but the starting materials have other ideas and form non-productive intermediates. Starting over, palladium catalysis proceeds smoothly, then relatively gentle oxidation (DDQ) produces the fully oxidized bruceolline E (see picture).
To access the final compound, Gribble & Co. must reduce just one of E's two ketones. The authors attempt borane reductions, but the "usual suspects" (CBS, Alpine) fail miserably. Optimization with (+)-DIP-Cl produces the final bruceolline J in high yield and ee. To make the unnatural enantiomer, the authors turn to a personal favorite: Baker's yeast, more commonly found in breads than labs. After 14 days in a warm, sweet slurry, the wee beasties return ent-bruceolline J in 98% ee.
The synthesis, only 4 short steps, should open the door to develop new antimalarial compounds.
*First, the Hawaii paradox - recruiting high-end, serious grad students to work 4-6 years in a tropical paradise. How does that work? And how do shipping delays from the mainland impact project selection? I can imagine that protecting group-free, relatively robust chemistry would have to be the norm, to survive storms, delays, humidity, etc.
Friday, August 16, 2013
Friday Fun: Super-Calcium!
 |
| Super-Calcium Source: Niggemann Group, RWTH Aachen |
A new trend almost snuck in under my radar: Calcium catalysis. In the past, a few groups had played around with amino-ene reactions, arylated tertiary alcohols, and made some enantioselective calcium pincer complexes. But I couldn't honestly tell you that I had branded any specific group with the "calcium" label, as opposed to the "palladium" or "organocatalysis" badges worn by many.
Well, the Niggemann group in Aachen, Germany appears to want that distinction. Prof. Meike and her team have released a slew of interesting reactions - Friedel / Crafts, [3+2] cyclizations, cyclopropanations - with more popping up seemingly monthly. But...calcium? The stuff ingrained in our bodies, stapled in the phosphate matrix of our bones and teeth? The stuff I eat in yogurt, milk, and cheese is now a catalyst?
 |
| Source: Niggemann Group, RWTH Aachen |
This new catalyst combo, dubbed "Super-Calcium" (with mascot, above), reacts like a wild hybrid of alumnium, gold, and palladium. It activates alcohols as leaving groups (Al). It permits [1,2] hydride shifts (Pd). It's a hard enough Lewis acid to unzip donor-acceptor cyclopropanes, but soft enough to permit hydroarylation (Al / Au). Checking some of the historical calcium-catalysis reactions (above) reveals even more head-scratching reactivity reminiscent of magnesium, titanium, or vanadium.
So, what's really going on here? First, I'd say it's early days: Some deuterium-labeling studies were done on the older reactions, and molecular modeling on this latest batch, but several steps (Vinyl cations? Hydride shifts?) make me wonder exactly how intimately the central calcium atom gets involved. Second, no one yet knows the exact structures of these reagents in solution; look how long it took to figure out LDA!!! Third, Meike's battle cry rings mostly true: reactions exploring the reactivity of early alkali metals (potassium? barium?) remain largely terra incognita.
More reactions will lead to more interest; perhaps a Calcium Craze looms over the horizon? Time will tell.
Happy Friday, Everyone!
-SAO
Thursday, July 18, 2013
"Crystalline" CO - Saccharin's New Trick
Seems I'm posting an awful lot about artificial sweeteners these days!
This latest example, from the always entertaining and informative folks at Angewandte Chemie, employs our old friend N-formylsaccharin* as a carbon monoxide "equivalent" for Pd-catalyzed addition reactions:
Quite a few benefits accrue: No pressure equipment. 'Medium' heat. Relatively low catalyst loading. Cheap, off-shelf reagents. If they could figure out a slightly less exotic reductant, I'd use this all the time!
This latest example, from the always entertaining and informative folks at Angewandte Chemie, employs our old friend N-formylsaccharin* as a carbon monoxide "equivalent" for Pd-catalyzed addition reactions:
 |
| Credit: Manabe Group | ACIEE |
Quite a few benefits accrue: No pressure equipment. 'Medium' heat. Relatively low catalyst loading. Cheap, off-shelf reagents. If they could figure out a slightly less exotic reductant, I'd use this all the time!
So, how does it work, anyway? The researchers confirm CO release by treating formylsaccharin with several bases and observing CO evolution.** The standard (boring) formylation model might be operative here - Pd oxidative addition, CO insertion, reductive cleavage (rinse, repeat).
OR (more excitingly), the authors note that they detect a transient "acylsaccharin" by HRMS. This might imply that the formylsaccharin reacts directly with the palladated arene, or that the sodium saccharine byproduct plays a role in stabilizing / promoting reduction of the insertion intermediate.
Sweet.
*OK, so it's not saccharin itself, but pretty darn close. First developed by Cossy in 2011 for formylating amines.
**A great mechanism for those looking for cume questions!
*OK, so it's not saccharin itself, but pretty darn close. First developed by Cossy in 2011 for formylating amines.
**A great mechanism for those looking for cume questions!
Saturday, June 8, 2013
Pyramid Power!
Atop the energy barrier between hardcore synthetic jocks and dyed-in-the-wool physical chemists lives a dedicated group: the molecular architects. A craving for perfect Platonic shapes, combined with the need to push the limits of bond and orbital theory, drive them towards projects like bullvalenes, cubane, or buckyballs. Despite the obvious challenges - bullvalenes shift and change, cubanes feel some strain - the structures' aesthetic appeal keeps us coming back for more.
Well, another class of shapely compounds seems poised to fall. Enter Pyramidanes*:
Reported a few days back in JACS by a collaboration between Japanese, French, and Russian scientists, the near-tetrahedral compounds are within shouting distance of an all-carbon version, never synthesized by human hands. I gotta say, they look pretty awesome. Just staring at it brings up so many questions: what's the hybridization of that top atom? How much strain energy? Could you make some schnazzy ligands from it? How labile is that Sn, anyway? Does this thing blow apart in TBAF?
The crew provides an in-depth discussion of frontier MO theory**, bond orders (fractional all around,
from 0.4-0.7), and NMR properties, concluding that there's a decent structural contribution from the ionic resonance form (see below).

The authors even suggest that the "cyclobutadiene" moiety - the pyramid "base" - can be stuck onto other metals; stable pyramidanes might work as strained-ring storage capsules.
Now I'm excited for the all-carbon version, which (theory predicts) has a lone pair at the apex...crazy!
---
*For the absurdly technical or IUPAC-minded, the authors offer alternative names such as tetracyclo-[2.1.0.0.0] pentane, or (my favorite) [3.3.3.3]fenestrane
**N.B. I'm no p-chemist, so someone else can go through the nitty-gritty there...
Well, another class of shapely compounds seems poised to fall. Enter Pyramidanes*:
 |
| Source: JACS | Lee, Sekiguchi |
The crew provides an in-depth discussion of frontier MO theory**, bond orders (fractional all around,
from 0.4-0.7), and NMR properties, concluding that there's a decent structural contribution from the ionic resonance form (see below).

The authors even suggest that the "cyclobutadiene" moiety - the pyramid "base" - can be stuck onto other metals; stable pyramidanes might work as strained-ring storage capsules.
Now I'm excited for the all-carbon version, which (theory predicts) has a lone pair at the apex...crazy!
---
*For the absurdly technical or IUPAC-minded, the authors offer alternative names such as tetracyclo-[2.1.0.0.0] pentane, or (my favorite) [3.3.3.3]fenestrane
**N.B. I'm no p-chemist, so someone else can go through the nitty-gritty there...
Wednesday, April 17, 2013
Precious Serendipity
Serendipity often drives scientific inquiry.
Ask a famous scientist - Feynman, Archimedes, Fleming, to name a few beneficiaries - they'd tell you it's better to be lucky than good. But Pasteur's paraphrased "Fortune favors the prepared mind" spells things out more clearly; diving into the nitty-gritty of chemical transformations often brings up a chance pearl of wisdom.
Such is the case for a recent JACS ASAP, out of the Wang group at Xiamen University in China. Let's set the stage: the scientists attempt to create a stabilized bimetallic cluster compound from gold, silver, and hemilabile P-N ligands. When the starting Ag-Au complex meets acetonitrile and methanol, something interesting occurs: a dimeric crystalline compound forms, bridged by two fully deprotonated acetonitriles!
So, why am I so excited?
1. That's a CCN (3-) anion stuck in there! What if you could, say, build a tetra-substituted carbon center just by adding electrophiles in sequence?
2. C-H activation of sp3 bonds = hard. Especially at room temp.
Well, there's (technically) 6 sp3 C-H bonds missing in that complex!
3. The authors note that water forms as the reaction progresses. Most nitriles, in the presence of Au/Ag catalysts and water, will hydrate to form amides. Not so here.
4. The authors point out that a gold oxo intermediate must be present for the cluster to form. They also point out that similar gold oxo catalysts (and silver oxide bases) pop up in the literature. Perhaps crazy intermediates like this commonly arise in gold-catalyzed reactions?
The best part? Studies like this always leave you with more questions than answers.
Perfect fodder for future projects.
Ask a famous scientist - Feynman, Archimedes, Fleming, to name a few beneficiaries - they'd tell you it's better to be lucky than good. But Pasteur's paraphrased "Fortune favors the prepared mind" spells things out more clearly; diving into the nitty-gritty of chemical transformations often brings up a chance pearl of wisdom.
Such is the case for a recent JACS ASAP, out of the Wang group at Xiamen University in China. Let's set the stage: the scientists attempt to create a stabilized bimetallic cluster compound from gold, silver, and hemilabile P-N ligands. When the starting Ag-Au complex meets acetonitrile and methanol, something interesting occurs: a dimeric crystalline compound forms, bridged by two fully deprotonated acetonitriles!
 |
| Those golden pyramids are pretty schnazzy. Source: JACS | Wang group |
1. That's a CCN (3-) anion stuck in there! What if you could, say, build a tetra-substituted carbon center just by adding electrophiles in sequence?
2. C-H activation of sp3 bonds = hard. Especially at room temp.
Well, there's (technically) 6 sp3 C-H bonds missing in that complex!
3. The authors note that water forms as the reaction progresses. Most nitriles, in the presence of Au/Ag catalysts and water, will hydrate to form amides. Not so here.
4. The authors point out that a gold oxo intermediate must be present for the cluster to form. They also point out that similar gold oxo catalysts (and silver oxide bases) pop up in the literature. Perhaps crazy intermediates like this commonly arise in gold-catalyzed reactions?
The best part? Studies like this always leave you with more questions than answers.
Perfect fodder for future projects.
Tuesday, February 12, 2013
Elsewhere...

Hey, check it out: Blog Syn #002 is live!
(And I hear that #003 is well on its way...)
More to come at JLC in the next few days. Stay tuned!
Friday, February 1, 2013
Pd Bites Back! Isopropyl Surprise
Although we don't always admit it, we chemists love a good surprise.
That's the feeling I got reading through a recent JACS ASAP, from the Chen group at PSU. They've been playing with an easy-to-remove C-H activation auxiliary (PA) first developed by Daugulis back in 2005. They've taught it some pretty neat tricks thus far: C-H amination, alkoxylation, and even alkylation reactions using simple primary iodides.
Their latest extends the chemistry to methyl iodide, which, when combined with some silver salts and a phosphate additive, usually plunks a new CH3 on an unhindered, kinetically-accessible gamma-methyl (i.e. the chain grows by one). However, in the case of norbornene, something wild happens: the Pd catalyst activates a secondary C-H bond, sticks a methyl on, but doesn't stop there; it "bites" into the newly formed methyl and adds on two more, to produce an isopropyl group. Sweet!
Chen's group shows that the position of the new i-Pr depends upon exo ("up") or endo ("down") relationships of the initial amine, but doesn't speculate much on mechanism.
Well, allow me! I wonder what happens if you keep feeding iodomethane into the system. I'm sure there's a limit (likely steric) that prohibits additional branching past a certain point, but currently nothing completely rules out a dendrimeric "norbornene tree" compound, right?
Readers, can someone disabuse me of this crazy notion?
That's the feeling I got reading through a recent JACS ASAP, from the Chen group at PSU. They've been playing with an easy-to-remove C-H activation auxiliary (PA) first developed by Daugulis back in 2005. They've taught it some pretty neat tricks thus far: C-H amination, alkoxylation, and even alkylation reactions using simple primary iodides.
Their latest extends the chemistry to methyl iodide, which, when combined with some silver salts and a phosphate additive, usually plunks a new CH3 on an unhindered, kinetically-accessible gamma-methyl (i.e. the chain grows by one). However, in the case of norbornene, something wild happens: the Pd catalyst activates a secondary C-H bond, sticks a methyl on, but doesn't stop there; it "bites" into the newly formed methyl and adds on two more, to produce an isopropyl group. Sweet!
 |
| Source: JACS ASAP 2013 | Chen group, PSU |
 |
| O Norbornene Tree, O Norbornene Tree; I do not grok your sterics. |
Readers, can someone disabuse me of this crazy notion?
Thursday, October 11, 2012
Cutting-Edge, Nobel-Worthy Chemistry
After all the early fuss about the merits of the 2012 Chemistry Nobel Prize, I noticed this challenge, couched in an earlier Chemjobber comment thread:
1. Whither Polymers? Darlings of early 20th-century industry, yet they've taken a back burner lately, winning their most recent Nobel in 2000. But, what a decade! Self-healing polymers. Fluoroelastomers you can print into any shape. Synthetic organs, even, grown from biodegradable polymer scaffolds. Trouble with this prize? Picking only three winners...
2. Biochemical Assembly Lines. Yes, cue the "it's not chemistry!" complaints, but I really like work which elucidates the cellular mechanisms plants, animals, and microbes use to assemble huge, medicinally-relevant natural products. Researchers can prompt E. coli to make an antifungal compound, for instance, or yeast to make a cancer therapy. Directed evolution of these assembly proteins, or the DNA which encodes them, can lead to products with wild substitutions and unexpected properties. Bonus: All the 'big wheels' tend to be card-carrying chemists, and work in chemistry departments. The overarching goal tends to be chemical - utilization of Nature's machinery to produce new compounds.
Usual suspects: Christopher Walsh, Chaitan Khosla, David Liu, Ben Shen.
3. Fundamental Catalysis. Technically, there have been a few Nobels for this fairly recently (2001, 2005, 2011). But, what a decade! Here's some currently-exploding fields:
Organocatalysis
Chiral Anion Catalysis
Gold Catalysis
New carbene ligands
Frustrated Lewis pairs
Catalytic C-H activation
Any discipline on this short list could take home a Nobel within 10 years. Admittedly, some of these are rather young, but, as Ash has pointed out, the committee has rewarded ever-shorter publication-to-prize gaps, so it's not without precedent.
Usual Suspects: Dean Toste, Melanie Sanford, Anthony Arduengo, Graham Hutchings, Douglas Stephan, David MacMillan, Benjamin List
Readers, who would you award a Chemistry Nobel?
"The organic chemists seem to get their hides chapped most easily when a Nobel gets awarded to a 'biologist'. It's worth asking 'what are the fundamental unanswered questions in organic chemistry?'" (Emphasis mine)Here are three areas, broadly defined, that I believe could win the Chemistry prize next year.
 |
| Synthetic trachea University College London, 2011 |
2. Biochemical Assembly Lines. Yes, cue the "it's not chemistry!" complaints, but I really like work which elucidates the cellular mechanisms plants, animals, and microbes use to assemble huge, medicinally-relevant natural products. Researchers can prompt E. coli to make an antifungal compound, for instance, or yeast to make a cancer therapy. Directed evolution of these assembly proteins, or the DNA which encodes them, can lead to products with wild substitutions and unexpected properties. Bonus: All the 'big wheels' tend to be card-carrying chemists, and work in chemistry departments. The overarching goal tends to be chemical - utilization of Nature's machinery to produce new compounds.
Usual suspects: Christopher Walsh, Chaitan Khosla, David Liu, Ben Shen.
 |
| Walsh Group, JACS 2012 |
3. Fundamental Catalysis. Technically, there have been a few Nobels for this fairly recently (2001, 2005, 2011). But, what a decade! Here's some currently-exploding fields:
Organocatalysis
Chiral Anion Catalysis
Gold Catalysis
New carbene ligands
Frustrated Lewis pairs
Catalytic C-H activation
Any discipline on this short list could take home a Nobel within 10 years. Admittedly, some of these are rather young, but, as Ash has pointed out, the committee has rewarded ever-shorter publication-to-prize gaps, so it's not without precedent.
Usual Suspects: Dean Toste, Melanie Sanford, Anthony Arduengo, Graham Hutchings, Douglas Stephan, David MacMillan, Benjamin List
Readers, who would you award a Chemistry Nobel?
Thursday, September 27, 2012
Methylamine Pseudoscience
Please see updates, posted below original piece. Thanks!
Pseudoscience strikes again. About a month ago, over at Slate's Brow Beat culture blog, Mr. Daniel Lametti - he of 'Ph.D.-Waste-Of-Time' fame - wrote a piece analyzing a recurring Breaking Bad plot device: the theft of large quantities of methylamine for the characters' illegal methamphetamine operation. The meat of the post:
As a practicing synthetic organic chemist, I agree with the statement that silica gel dehydrates solvents by water absorption. Sure. But I've heated plenty of alcohols in the presence of silica gel, and 99% of them don't spontaneously lose water! (That would be a rocking olefin synthesis, if it worked...)
Let's put this on firm scientific ground. The reaction in question, a nucleophilic substitution, could theoretically occur by two mechanisms: SN1, where the -OH group of methanol dislodges to form a methyl cation (?!?!), followed by subsequent ammonia bonding; or SN2, where the ammonia directly displaces the -OH, one step, no intermediates.
In this scenario, both are extremely unlikely, especially at room temperature and pressure.
Now let's talk practicality: which company will sell you a cylinder of ammonia gas for 'home use?' (Not Home Depot). How will you get your methanol? What's the plan to isolate the (volatile, stinky) methylamine from the complex mixture of compounds this theoretical reaction produces?
Well, how do companies make methylamine? Albemarle technical documents to the rescue! Seems that mixing methanol and excess ammonia at 300-500 degrees Celsius, under pressure, over a zeolite catalyst will produce an equilibrium mixture of methylamine, dimethylamine, and trimethylamine (favored). After fractional distillation, the trimethylamine can be streamed over an amorphous silica / alumina catalyst to disproportionate it back into methylamine.
Not a kitchen sink in sight.
Update, 8/17/13 - This piece jumped back into the spotlight as Breaking Bad winds down its 5-season run. Thanks to Dylan at WaPo's Wonkblog for linking back here.
Commenters have taken issue with my description of the reaction, so I've slightly changed the text for clarity (methods / mechanisms, "forms" cations, oxidation...)
8/19/13 - Arguments have cropped up, in multiple forums, about reagent availability, feasibility of the chemistry at small-scale, mechanism, purification, etc. I blame myself for not refining my argument well enough in the original post. Very directly, I'll re-state the major arguments:
1) The silica gel + methanol + ammonia route will not produce methylamine as stated
2) Although an experienced chemist *could* produce methylamine using different reactions in a kitchen sink, he will by no means produce enough to support a burgeoning criminal enterprise which manufactures methamphetamine at multi-kilo scale.
*Curious - Appended at the bottom of the essay is a thanks for Prof. Adam Braunschweig, facultyat NYU now UMiami.. To what extent did Prof. Braunschweig proofread this post? Did he sign off on the "kitchen sink silica gel" concept in the middle? I can't possibly imagine that he thoroughly vetted this essay.
Pseudoscience strikes again. About a month ago, over at Slate's Brow Beat culture blog, Mr. Daniel Lametti - he of 'Ph.D.-Waste-Of-Time' fame - wrote a piece analyzing a recurring Breaking Bad plot device: the theft of large quantities of methylamine for the characters' illegal methamphetamine operation. The meat of the post:
Wait, huh? Let's start from No."As a post on Reddit asks, since Walt is a brilliant chemist, couldn’t he just synthesize the stuff himself?Yes, and pretty easily. There are many different ways to make the compound; with little more than an introductory organic chemistry class, you could probably synthesize it in your kitchen sink. (Brow Beat doesn’t recommend trying to make methylamine in your kitchen sink). Chemically speaking, methylamine is just ammonia with one hydrogen atom swapped out for a methyl group—a carbon atom and three hydrogen atoms. Without getting into too much detail, an easy way to achieve this swap* is to “bubble” ammonia (a gas) through methanol (a liquid) that’s been laced with a dehydrating agent like Silica gel. You could probably buy these chemicals at Home Depot and CVS. Silica gel packets are often packaged with new shoes and electronics to keep them dry."
As a practicing synthetic organic chemist, I agree with the statement that silica gel dehydrates solvents by water absorption. Sure. But I've heated plenty of alcohols in the presence of silica gel, and 99% of them don't spontaneously lose water! (That would be a rocking olefin synthesis, if it worked...)
 |
| Methylamine: Easy as hooking up five pressurized reactors in your kitchen sink. Credit: Albermarle |
In this scenario, both are extremely unlikely, especially at room temperature and pressure.
Now let's talk practicality: which company will sell you a cylinder of ammonia gas for 'home use?' (Not Home Depot). How will you get your methanol? What's the plan to isolate the (volatile, stinky) methylamine from the complex mixture of compounds this theoretical reaction produces?
Well, how do companies make methylamine? Albemarle technical documents to the rescue! Seems that mixing methanol and excess ammonia at 300-500 degrees Celsius, under pressure, over a zeolite catalyst will produce an equilibrium mixture of methylamine, dimethylamine, and trimethylamine (favored). After fractional distillation, the trimethylamine can be streamed over an amorphous silica / alumina catalyst to disproportionate it back into methylamine.
Not a kitchen sink in sight.
Update, 8/17/13 - This piece jumped back into the spotlight as Breaking Bad winds down its 5-season run. Thanks to Dylan at WaPo's Wonkblog for linking back here.
Commenters have taken issue with my description of the reaction, so I've slightly changed the text for clarity (methods / mechanisms, "forms" cations, oxidation...)
8/19/13 - Arguments have cropped up, in multiple forums, about reagent availability, feasibility of the chemistry at small-scale, mechanism, purification, etc. I blame myself for not refining my argument well enough in the original post. Very directly, I'll re-state the major arguments:
1) The silica gel + methanol + ammonia route will not produce methylamine as stated
2) Although an experienced chemist *could* produce methylamine using different reactions in a kitchen sink, he will by no means produce enough to support a burgeoning criminal enterprise which manufactures methamphetamine at multi-kilo scale.
*Curious - Appended at the bottom of the essay is a thanks for Prof. Adam Braunschweig, faculty
Wednesday, July 25, 2012
Great Expectations for 'Metal-Free' Reactions
Reading through some recent "metal-free" coupling literature, I came across a fantastic footnote. Check out the lengths chemists Carsten Bolm and Isabelle Thomé have to go to in order to certify their latest reaction:
Wow.
I'll be honest with you, I've never tested for metals in my starting materials below ~1 ppm (Food for thought: here's an EMA document detailing allowable catalyst residue in human medicines). I'd wager that 99.9% of workaday bench chemists haven't, either.
Bolm's group endures this rigor because, well, they literally wrote the book on trace metal catalysis. Quite honestly, I'd bet that they felt a bit uneasy when they measured the DMEDA copper concentration; more than a few of these "metal-free" reactions proceed with vanishingly small amounts of catalyst.
"(16) Great care was taken to avoid the presence of transition metal impurities. All starting materials were synthesized without using any transition metal...reagent transfers were performed with one-way plastic spatulas, and new glassware and stirbars were used for the cyclization reactions. The starting materials and reagents were analyzed to the detection limit of 4 ppb by atomic absorption spectroscopy (AAS) or inductively coupled mass spectrometry (ICP-MS).
Data [for a representative intermediate] - Cu < 4ppb, Pd <4 ppb; Kcarb - Cu < 4 ppb, Pd < 4 ppb; DMEDA [ligand] - Cu 2.4 ppm, Pd < 4 ppb." [Emphasis mine]
 |
| Sand - Probably > 4ppb "active" metals! Source: 123RF |
I'll be honest with you, I've never tested for metals in my starting materials below ~1 ppm (Food for thought: here's an EMA document detailing allowable catalyst residue in human medicines). I'd wager that 99.9% of workaday bench chemists haven't, either.
Bolm's group endures this rigor because, well, they literally wrote the book on trace metal catalysis. Quite honestly, I'd bet that they felt a bit uneasy when they measured the DMEDA copper concentration; more than a few of these "metal-free" reactions proceed with vanishingly small amounts of catalyst.
Saturday, March 24, 2012
Chemistry "Hacks" (The Good Kind)
 |
| My hackathon drink of choice, coffee! (And hey, ACS, why no more mugs?) |
Let's go all the way back to the beginning of chemical research - who were we? Alchemists, who worked after hours, scribbling in secret languages; some hoped for profit, and some just loved the thrill (Sound familiar?). They didn't follow the implicit hegemony we do today: school student -> university trainee -> graduate study -> postdoc -> junior professor -> original ideas? By the time you're done jumping through hoops, you might have left your sense of curiosity and wonder behind.
 |
| Early Chemical "Hacker" Alchemist with Scale, Johannes Weiland Credit: Chemical Heritage Foundation |
Ever beat your head against a research problem, only to find the answer at a neighboring department's seminar? Borrowed something the next lab down the hall had on the shelf? Not to wax all Zen and the Art of Motorcycle Maintenance on you, but I subscribe to human quality precognition, a subtle mental undercurrent that guides you toward the right reaction or correct conditions. Others might call this gut instinct, and I've heard process chemists chat about initial optimization ("lucky on the first try!").
So why not speed this process along? Could we take a page from the video game designers, the hackers, and the dorm-room dot-com stars?
Here's my proposal (which, incidentally, might work pretty well at a large, national chemistry conference, just sayin'): What if visiting chemists had access to an open lab space, replete with all the latest catalysts, equipment, and reagents? One could imagine equipment dealers sponsoring this space, much like Cuisinart and KitchenAid sponsor cooking shows.
 |
| Picture this, but with more fume hoods and Buchwald ligands |
Who knows? Maybe, in time, the phrase "chemistry hack" might mean something good!
Tuesday, February 28, 2012
The 2011 Organometallics Roundtable – Peering into the Future
(Note: I’m publishing this post concurrently with my blog bud Chemjobber. Hop on over to his site to read about the industry / academia training and #chemjobs angles. This way, regular readers get twice the opinions for half the price!)
 |
| Who wants to chat? Source: Texas A&M U. |
As 2011 drew to a close, John Gladysz, the new Chief Editor of Organometallics, sat down for a chat with seventeen organometallic chemists from different national (German, Swiss, Australian, US, Chinese, British) and employment backgrounds (14 academia, 2 industrial, 1 government). The result? An in-depth discussion, full of banter and back-talk, which covers topics such as industrial training for grad students, national creativity differences, “dream reactions,” and how to encourage industrial cross-collaboration.
As an icebreaker, Gladysz had each chemist dream up their “Christmas Stocking Reaction,” the ultimate goal each would love to see realized. Here’s a quick rundown:
- Pragmatism –Joachim Ritter (DuPont) envisioned several scenarios for taking non-petroleum-derived feedstocks on to commodity chemicals. Ritter’s focus on hydroxymethyl-furfural (HMF, a darling of ChemEng labs across the country) was perhaps unsurprising, but I enjoyed his new ideas for deoxygenation of vegetable oils to produce fine chemicals. Jerzy Klosin (Dow) chimed in for development of newer, cheaper catalytic complexes, especially championing first-row analogues for replacement of palladium in polymerizations. Bernhard Rieger (Technische U. Munschen) weighed in on incorporation of CO2 into polymers with nickel catalysts.
- Break the Glove Boxes! – Jim Mayer (U. Washington) and Bill Jones (U. Rochester), building off of Ritter’s theoretical deoxygenation catalysts, proposed chemistry based on high-oxidation-state metals. Mark Humphrey (Australia National) wished for an air-insensitive route to Sonogoshira-type couplings of alkynyl dendrimers. Jennifer Schomaker (U. Wisc.-Madison) hoped to use nitrous oxide as a terminal oxidant, and Vy Dong (U. of Toronto) would like chiral ligands for high-valent Pd chemistry.
 |
| P-N-N pincers and Pd oxidation, Z-metathesis and dehydrogenation, Iridium cat'lysts that stitch up new rings These are a few of their favorite things... |
- Mind your MOFs – Still others, such as Zach Ball (Rice U.) and Ekkehardt Hahn (Westfalische Wilhelms-Universitat Munster) weighed in on new types of building blocks and metals for cluster and MOF chemistry. Vivian Yam (U. of Hong Kong) wrapped up everyone’s requests with a nice bow; she hoped for new air- and moisture-insensitive OLED materials, solar energy-collecting polymers, and water-splitting photocatalysts.
Filling the “Tool Box” - Rieger cautioned to watch out for the recurring industrial opinion that “all the useful chemistry is already discovered.” Gladysz used the example of frustrated Lewis pairs, which, while a fairly young concept, are already turning heads. Bill Jones gave an impromptu one-liner about heterolytic hydrogenation: “If someone had written that on an exam…you’d give them a zero (back in the old days).” Humphrey chimed in on bimetallic bases, and Klosin for Ziegler-Natta studies.
But just then, Ritter poured cold water on the party: “Today’s chemical companies are busy reacting to rapid market swings and trends, which does not leave a lot of room for risky long-term projects.” Sensing, perhaps, that he’d deflated the roundtable’s enthusiasm, he quickly backtracked and mentioned that he’s “looking forward to…CO2 utilization, solar energy, and biomass based energy and chemicals.”
So there you go, future faculty members, he’s written half your proposal already!
Throwaway lines – CJ has collected quite a few of these, but I’d like to comment on a few more.
“I’ve talked to a colleague in China whose professor advised ‘If you don’t do palladium chemistry, how will people know you are my student?’.” –Vy Dong
 |
| Peering into the OM "xstal ball " Source: Organometallics |
This creativity discussion wound through the group, until Yam and Hahn debated whether Indian and Chinese universities still seemed to hire based on quantity (i.e. papers published) vs. quality.
Hahn: “…even the 28-year old researchers from China know their h-indexes [and] introduce themselves saying their name and ‘I have an h-index of 10.’ And you wonder who has trained them?”
I can’t say that I’ve never encountered the "publish or die" sentiment, but I had hoped it was becoming less prevalent with time. Readers, your thoughts?
The Last Word - “…in my opinion, the chemical community performs poorly in transmitting to the general public what they are actually doing.” –Ekke relays a sobering thought for those of us in the blogosphere who toil daily to demystify our science.
Maybe he doesn’t read enough good science blogs?
Subscribe to:
Posts (Atom)